Concours de vulgarisation scientifique 2015 | finaliste
La danse des molécules : une façon d’accélérer la découverte de médicaments
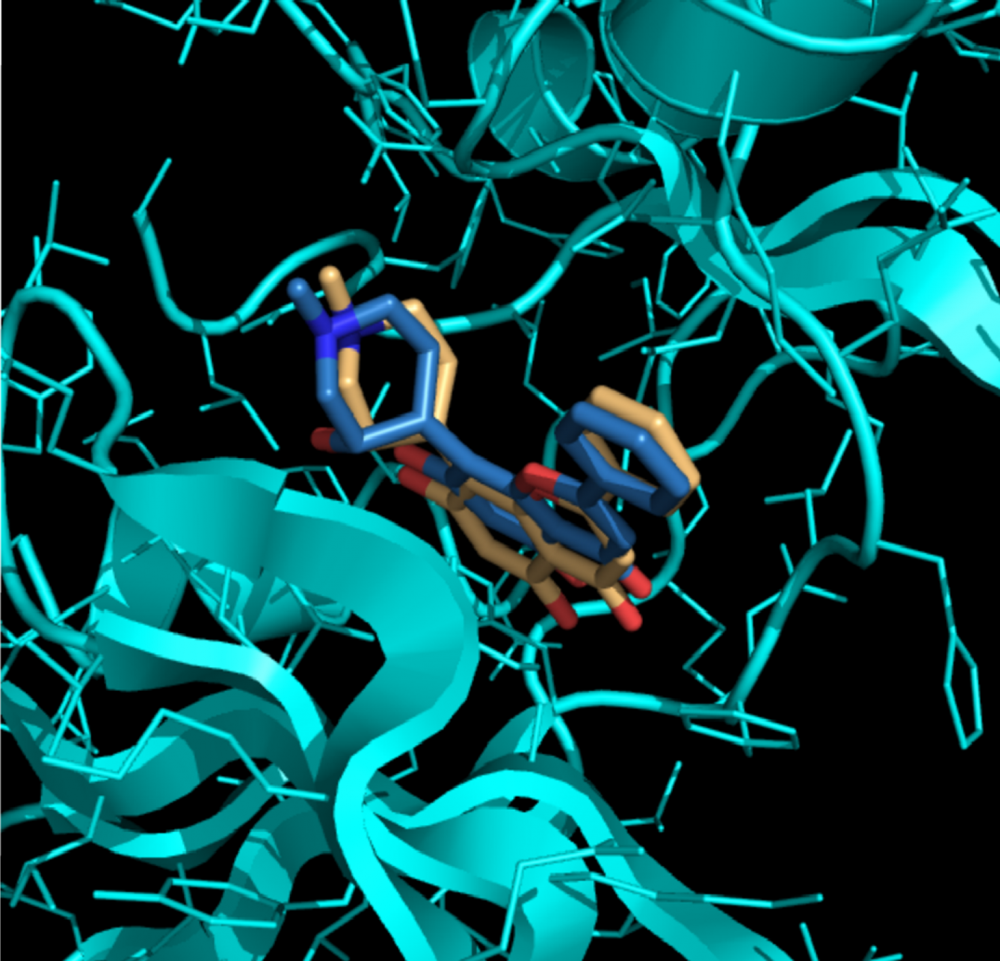
Photo : Image fournie par Mathieu Larocque
Une célèbre chanson latino-américaine dit que « la vie est une danse et le monde, une piste de danse ». Il s'agit également d'une expression populaire que les grands-mères se plaisent souvent à citer. À vrai dire, on pourrait presque appliquer cette citation aux interactions entre les entités biologiques.
Plus précisément, à l'échelle moléculaire, les protéines, éléments constitutifs de notre organisme au même titre que d'autres molécules, s’opposent aux autres molécules dans une danse perpétuelle. Concrètement, on peut observer cette danse dans la majorité des processus cellulaires et biochimiques qui se produisent à l'intérieur d'une cellule.
Comme c'est le cas pour les humains, ce ne sont pas toutes les molécules qui parviennent à danser sur n'importe quel rythme. De plus, certaines d'entre elles sont extrêmement sélectives quant à leur choix de partenaire de danse. Afin de déterminer s'il existe une interaction particulière entre une protéine et une autre molécule, les scientifiques disposent d'une gamme complète d'outils appartenant à la biophysique, la biologie structurale et la bio-informatique structurale.
Les techniques biophysiques sont très développées et trop nombreuses pour en faire l’énumération ici, elles offrent la possibilité de mesurer parfois précisément l’affinité de liaison d’une petite molécule à une protéíne. La biologie structurale s'applique à représenter de manière concrète des phénomènes biologiques qui existent à très petite échelle, soit à l'échelle moléculaire et atomique. L'objectif principal de cette branche de la biologie est de comprendre les formes, les structures et les interactions des molécules à l'origine de l'ensemble des processus biologiques qui se produisent dans les cellules. Pour ce faire, les scientifiques ont recours à des techniques de pointe comme la diffraction des rayons X, la résonance magnétique nucléaire (RMN) et plus récemment la microscopie cryoélectronique. Toutes ces techniques sont très importantes, particulièrement en recherche pharmaceutique, où le principal objectif consiste à sélectionner de nouveaux candidats-médicaments pour le traitement de différents troubles médicaux. Toutefois, leur mise en place est un processus long et coûteux.
Parmi les techniques de bio-informatique structurale, l’arrimage moléculaire fait son entrée en scène en tant qu'importante méthode d'identification de nouveaux médicaments. Cette technique vise à prédire la position et l'orientation d'une molécule (le ligand) par rapport à la représentation virtuelle de la structure tridimensionelle d’une protéine (dont la fonction biologique est importante, par exemple une enzyme). En recherche médicale, on emploie principalement cette méthode pour le criblage de médicaments et de substances chimiques faisant partie de grosses bases de données. En d'autres mots, on simule l'arrimage de ces derniers avec une série de petites molécules différentes et on ne retient que les candidats les plus prometteurs. Le but des scientifiques consiste donc à déterminer quels « danseurs » ont les plus fortes affinités. Les candidats retenus font par la suite l'objet d'essais expérimentaux en laboratoire.
Il existe une pléthore de logiciels et de serveurs Web conçus pour l'arrimage moléculaire qui permettent d'effectuer ce criblage virtuel. Néanmoins, la plupart de ces programmes n’ont pas d'interface utilisateur et chacun d'eux présente des limites techniques qui peuvent brouiller et compliquer les analyses (et c'est encore plus vrai si le chercheur ne connaît pas les bases de la bio-informatique). Le groupe de recherche Najmanovich (un groupe de recherche de la Faculté de médecine et des sciences de la santé de l'Université de Sherbrooke) est conscient des limites des non-initiés en ce qui a trait à la biologie structurale et à la modélisation moléculaire1. Dirigé par le Pr Rafael Najmanovich, le groupe a développé un logiciel qu'il a baptisé NRGsuite (annonce parue dans la revue Bioinformatics du mois d'août)2. NRGsuite permet aux scientifiques qui ne s'y connaissent pas en bio-informatique d'effectuer, d'une manière intuitive et conviviale l'arrimage moléculaire. Cette interface permet à l'utilisateur d'obtenir une représentation graphique en temps réel des analyses. Autrement dit, il est possible d'observer ce que l'on fait. Les scientifiques peuvent ainsi évaluer si la danse entre les molécules et les protéines se déroule sans heurts et au bon rythme. NRGsuite intègre notamment deux outils : GetCleft et FlexAID. Le premier permet de définir où les deux partenaires de danse se tiendraient la main (le site de liaison dans la protéine) lors d’une danse précise et le deuxième permet de vérifier comment ces danseurs le feront en déterminant la structure plus probable pour le complexe protéine/petite-molécule. Ces paramètres permettent de déterminer si ces « couples » peuvent réellement évoluer en parfaite harmonie sur la piste de danse3. NRGsuite fonctionne sous Windows, Linux et Mac OS.
« Le criblage virtuel permet de sélectionner plus rapidement des molécules et des médicaments. Mettre au point ce genre d'outil n'est pas chose facile. Il faut beaucoup de temps et d’habiletés en informatique. Toutefois, si on compare la technique d'arrimage moléculaire aux procédures expérimentales, l’arrimage moléculaire permet de gagner du temps car il est possible de tester plusieurs molécules virtuellement et éliminer celles qui n’ont aucune chance de succès. Les essais expérimentaux pourront cibler les molécules ayant le plus de potentiel. Qui plus est, dans notre cas, NRGsuite a été conçu en collaboration avec les étudiants selon une approche axée sur l'enseignement. Ainsi, après avoir recueilli pendant quatre ans les commentaires et les suggestions des étudiants, nous avons lancé le logiciel en sachant qu'il était adapté à tous les types de publics », affirme le Pr Najmanovich.
On peut notamment lire dans une étude publiée dans le Journal of Medicinal Chemistry par ce même groupe de recherche4, que des logiciels de simulation d'arrimage ont été utilisés dans le but de trouver de bons inhibiteurs (médicaments qui empêchent une réaction chimique de se produire) pour une protéine appelée la matriptase. Cette protéine intervient dans plusieurs processus cellulaires et semble jouer un rôle capital dans l'évolution de certaines maladies, comme l'arthrose, et certains types de tumeurs. Ces travaux de recherche ont permis d'identifier de nouveaux puissants inhibiteurs de la matriptase parmi 8 000 molécules de départ. « Cette technique n'est pas seulement importante dans le domaine de la recherche de nouveaux médicaments. Si les chercheurs veulent comprendre comment une molécule donnée interagit ou découvrir l'endroit où se produit une interaction, la technique d'arrimage moléculaire pourrait orienter leur prise de décisions concernant les études expérimentales », ajoute Mathieu Larocque, un étudiant aux cycles supérieurs membre du groupe de recherche.
Comme c'est le cas dans la vraie vie, on ne trouve pas notre partenaire idéal en claquant des doigts. Toutefois, il faut de la pratique pour être bon danseur, et les scientifiques l'ont compris. Un nombre croissant de molécules sont testées quotidiennement, et aujourd'hui, grâce aux outils de plus en plus précis et à l'accessibilité grandissante des logiciels, l'avenir de la santé humaine est prometteur. C'est ainsi que nous pourrons suivre l'évolution des maladies, vieilles ou nouvelles.
Références
1. NRG. Najmanovich Research Group. (2015). at <http://bcb.med.usherbrooke.ca/index.php>
2. Gaudreault, F., Morency, L.-P. & Najmanovich, R. J. NRGsuite: a PyMOL plugin to perform docking simulations in real time using FlexAID. Bioinformatics btv458– (2015). doi:10.1093/bioinformatics/btv458
3. Gaudreault, F. & Najmanovich, R. J. FlexAID: Revisiting Docking on Non-Native-Complex Structures. J. Chem. Inf. Model. 55, 1323–36 (2015).
4. Duchêne, D. et al. Analysis of subpocket selectivity and identification of potent selective inhibitors for matriptase and matriptase-2. J. Med. Chem. 57, 10198–204 (2014).